
GPS 2.0, a Tool to Predict Kinase-specific Phosphorylation Sites in Hierarchy
Molecular & Cellular Proteomics. 2008;7(9):1598-1608.
[
Abstract
]
[
Full Text
]
Read more
GPS 2.0
Identification of protein phosphorylation sites with their cognate protein kinases (PKs) is a key step to
delineate molecular dynamics and plasticity underlying a variety of cellular processes.
In this work, we adopted a well established rule to classify PKs into a hierarchical structure with four
levels, including group, family, subfamily, and single PK.
In addition, we developed a simple approach to estimate the theoretically maximal false positive rates.
The on-line service and local packages of the GPS (Group-based Prediction System) 2.0 were implemented in
Java with the modified version of the Group-based Phosphorylation Scoring algorithm. As the first stand
alone software for predicting phosphorylation, GPS 2.0 can predict kinase-specific phosphorylation sites for
408 human PKs in hierarchy. A large scale prediction of more than 13,000 mammalian phosphorylation sites by
GPS 2.0 was exhibited with great performance and remarkable accuracy.Thus, the GPS 2.0 is a useful tool for
predicting protein phosphorylation sites and their cognate kinases and is freely available on line.
GPS 2.0 now is updated as GPS3.0 and is freely available at
http://gps.biocuckoo.org.

CSS-Palm 2.0: an updated software for palmitoylation sites prediction
Protein Engineering, Design and Selection. 2008;21(11):639-644.
[
Abstract
]
[
Full Text
]
ESI HCP
Read more
CSS-Palm 2.0
Protein palmitoylation is an essential post-translational lipid modification of proteins, and reversibly
orchestrates a variety of cellular processes.
In this work, we updated our previous CSS-Palm into version 2.0. An updated clustering and scoring strategy
(CSS) algorithm was employed with great improvement.
The leave-one-out validation and 4-, 6-, 8- and 10-fold cross-validations were adopted to evaluate the
prediction performance of CSS-Palm 2.0.
Also, an additional new data set not included in training was used to test the robustness of CSS-Palm 2.0.
As an application, we performed a small-scale annotation of palmitoylated proteins in budding yeast.
The online service and local packages of CSS-Palm 2.0 were freely available at:
http://csspalm.biocuckoo.org

DOG 1.0: illustrator of protein domain structures
Cell Research. 2009;19(2):271-273.
[
Abstract
]
[
Full Text
]
Read more
DOG 1.0
Development of computer software that can illustrate user-designated protein domain structures will be a
great help for biological experimentalists to communicate their research results.
In this work, we present a novel software of DOG (Domain Graph, version 1.0) for experimentalists, to
prepare publication-quality figures of protein domain structures.
The scale of a protein domain and the position of a functional motif/site will be precisely defined.
The DOG 1.0 software was written in JAVA 1.5 (J2SE 5.0) and packed with Install4j 4.0.8.
Then we developed several packages to support three major Operating Systems (OS), including Windows,
Unix/Linux and Mac.
For Windows and Linux systems, a Java Runtime Environment 6 (JRE) package of Sun Microsystems was also
included.
The DOG 1.0 software is freely available from:
http://dog.biocuckoo.org.

Systematic study of protein sumoylation: Development of a site-specific predictor of SUMOsp 2.0
Proteomics. 2009;9(12):3409-3412.
[
Abstract
]
[
Full Text
]
Read more
SUMOsp 2.0
Protein sumoylation is an important reversible post-translational modification on proteins, and orchestrates
a variety of cellular processes.
In this work, we developed SUMOsp 2.0, an accurate computing program with an improved group-based
phosphorylation scoring algorithm.
Our analysis demonstrated that SUMOsp 2.0 has greater prediction accuracy than SUMOsp 1.0 and other existing
tools, with a sensitivity of 88.17% and a specificity of 92.69% under the medium threshold.
Previously, several large-scale experiments have identified a list of potential sumoylated substrates in
Saccharomyces cerevisiae and Homo sapiens;
however, the exact sumoylation sites in most of these proteins remain elusive. We have predicted potential
sumoylation sites in these proteins using SUMOsp 2.0,
which provides a great resource for researchers and an outline for further mechanistic studies of
sumoylation in cellular plasticity and dynamics.
The online service and local packages of SUMOsp 2.0 are freely available at:
http://sumosp.biocuckoo.org

MiCroKit 3.0: an integrated database of midbody, centrosome and kinetochore
Nucleic Acids Research. 2010;38:D155-D160.
[
Abstract
]
[
Full Text
]
Read more
MiCroKit 3.0
During cell division/mitosis, a specific subset of proteins is spatially and temporally assembled into
protein super complexes
in three distinct regions, i.e. centrosome/spindle pole, kinetochore/centromere and midbody/cleavage
furrow/phragmoplast/bud
neck, and modulates cell division process faithfully. Here, we present the MiCroKit database (
http://microkit.biocuckoo.org) of proteins that localize in
midbody, centrosome and/or kinetochore. We collected into the MiCroKit database experimentally
verified microkit proteins from the scientific literature that have unambiguous supportive evidence for
subcellular localization
under fluorescent microscope. The current version of MiCroKit 3.0 provides detailed information for 1489
microkit proteins
from seven model organisms, including
Saccharomyces cerevisiae,
Schizasaccharomyces pombe,
Caenorhabditis elegans,
Drosophila melanogaster,
Xenopus laevis,
Mus
musculus and
Homo sapiens. Moreover, the orthologous information was provided for these
microkit proteins, and could be a useful resource for further
experimental identification.

PhosSNP for Systematic Analysis of Genetic Polymorphisms That Influence Protein Phosphorylation
Molecular & Cellular Proteomics. 2010;9(4):623-634.
[
Abstract
]
[
Full Text
]
Read more
PhosSNP
We are entering the era of personalized genomics as breakthroughs in sequencing technology have made it
possible to sequence or genotype an individual person in an efficient and accurate manner.
Preliminary results from HapMap and other similar projects have revealed the existence of tremendous genetic
variations among world populations and among individuals.
It is also generally believed that the genetic variation is the main cause for different susceptibility to
certain diseases or different response to therapeutic treatments.
In this work, using an in-house developed kinase-specific phosphorylation site predictor (GPS 2.0), we
computationally detected that ∼70% of the reported nsSNPs are potential phosSNPs.
Finally, all phosSNPs were integrated into the PhosSNP 1.0 database, which was implemented in JAVA 1.5 (J2SE
5.0).
The PhosSNP 1.0 database is freely available for academic researchers at:
http://phossnp.biocuckoo.org

GPS-SNO: Computational Prediction of Protein S-Nitrosylation Sites with a Modified GPS Algorithm
Plos One. 2010;5(6): e11290.
[
Abstract
]
[
Full Text
]
Read more
GPS-SNO
As one of the most important and ubiquitous post-translational modifications (PTMs) of proteins,
S-nitrosylation
plays important roles in a variety of biological processes, including the regulation of cellular dynamics
and plasticity. Identification of
S-nitrosylated substrates with their exact sites is crucial for
understanding the molecular mechanisms of
S-nitrosylation.In this work, we developed a novel
software of GPS-SNO 1.0 for the prediction of
S-nitrosylation sites.By comparison, the prediction
performance of GPS 3.0 algorithm was better than other methods, with an accuracy of 75.80%, a sensitivity of
53.57% and a specificity of 80.14%. As an application of GPS-SNO 1.0, we predicted putative
S-nitrosylation
sites for hundreds of potentially
S-nitrosylated substrates for which the exact
S-nitrosylation
sites had not been experimentally determined.The online service and local packages of GPS-SNO were
implemented in JAVA and are freely available at:
http://sno.biocuckoo.org.

A Summary of Computational Resources for Protein Phosphorylation
Current Protein & Peptide Science. 2010;11(6):485-496.
[
Abstract
]
[
Full Text
]
Read more
Protein Phosphorylation
Protein phosphorylation is the most ubiquitous post-translational modification (PTM), and plays important
roles in most of biological processes. Identification of site-specific phosphorylated substrates is
fundamental for understanding the molecular mechanisms of phosphorylation. Besides experimental approaches,
prediction of potential candidates with computational methods has also attracted great attention for its
convenience, fast-speed and low-cost. In this review, we present a comprehensive but brief summarization of
computational resources of protein phosphorylation, including phosphorylation databases, prediction of
non-specific or organism-specific phosphorylation sites, prediction of kinase-specific phosphorylation sites
or phospho-binding motifs, and other tools. The latest compendium of computational resources for protein
phosphorylation is available at:
http://gps.biocuckoo.org/links.php
CPLA 1.0: an integrated database of protein lysine acetylation
Nucleic Acids Research. 2011;39:D1029-1034.
[
Abstract
]
[
Full
Text
]
Read more
CPLA 1.0
As a reversible post-translational modification (PTM) discovered decades ago, protein lysine acetylation was
known for its regulation of transcription through the modification of histones. Recent studies discovered
that lysine acetylation targets broad substrates and especially plays an essential role in cellular
metabolic regulation.In this work, we presented the compendium of protein lysine acetylation (CPLA) database
for lysine acetylated substrates with their sites. The online services of CPLA database
was implemented in PHP + MySQL + JavaScript, while the local packages were
developed in JAVA 1.5 (J2SE 5.0). The CPLA database
is updated as CPLM and is freely available for all users at:
http://cplm.biocuckoo.org

GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length
selection
Protein Engineering, Design and Selection. 2011;24(3):255-260.
[
Abstract
]
[
Full Text
]
Read more
GPS 2.1
As the most important post-translational modification of proteins, phosphorylation plays essential roles in
all aspects of biological processes. Besides experimental approaches, computational prediction of
phosphorylated proteins with their kinase-specific
phosphorylation sites has also emerged as a popular strategy, for its low-cost, fast-speed and convenience.
In this work,
we developed a kinase-specific phosphorylation sites predictor of GPS 2.1 (Group-based Prediction System),
with a novel but
simple approach of motif length selection (MLS). By this approach, the robustness of the prediction system
was greatly improved.
All algorithms in GPS old versions were also reserved and integrated in GPS 2.1. The online service and
local packages of
GPS 2.1 were implemented in JAVA 1.5 (J2SE 5.0) and freely available for academic researches at:
http://gps.biocuckoo.org

GPS-YNO2: computational prediction of tyrosine nitration sites in proteins
Mol. BioSyst. 2011;7(4):1197-1204.
[
Abstract
]
[
Full
Text
]
Read more
GPS-YNO2
The last decade has witnessed rapid progress in the identification of proteintyrosine nitration
(PTN), which is an essential and ubiquitous post-translational modification (PTM) that plays a variety of
important roles in both physiological and pathological processes, such as the immune response, cell death,
aging and neurodegeneration.
Identification of site-specific nitrated substrates is fundamental for understanding the molecular
mechanisms and biological functions of PTN.
In contrast with labor-intensive and time-consuming experimental approaches, here we report the development
of the novel software package GPS-YNO2 to predict PTN sites.
The software demonstrated a promising accuracy of 76.51%, a sensitivity of 50.09% and a specificity of
80.18% from the leave-one-out validation.
Through a statistical functional comparison with the nitric oxide (NO) dependent reversible modification of
S-nitrosylation, we observed that PTN prefers to attack certain fundamental biological processes and
functions.
Finally, the online service and local packages of GPS-YNO2 1.0 were implemented in JAVA and freely available
at:
http://yno2.biocuckoo.org
GPS-CCD: A Novel Computational Program for the Prediction of Calpain Cleavage Sites
Plos One. 2011;6(4):e19001.
[
Abstract
]
[
Full Text
]
Read more
GPS-CCD
As one of the most essential post-translational modifications (PTMs) of proteins, proteolysis, especially
calpain-mediated cleavage, plays an important role in many biological processes, including cell
death/apoptosis, cytoskeletal remodeling, and the cell cycle. Experimental identification of calpain targets
with
bona fide cleavage sites is fundamental for dissecting the molecular mechanisms and biological
roles of calpain cleavage. In contrast to time-consuming and labor-intensive experimental approaches,
computational prediction of calpain cleavage sites might more cheaply and readily provide useful information
for further experimental investigation. In this work, we constructed a novel software package of GPS-CCD
(Calpain Cleavage Detector) for the prediction of calpain cleavage sites, with an accuracy of 89.98%,
sensitivity of 60.87% and specificity of 90.07%. With this software, we annotated potential calpain cleavage
sites for hundreds of calpain substrates, for which the exact cleavage sites had not been previously
determined.The online service and local packages of GPS-CCD 1.0 were implemented in JAVA and are freely
available at:
http://ccd.biocuckoo.org/.
GPS-PUP: computational prediction of pupylation sites in prokaryotic proteins
Mol. BioSyst. 2011;7(10):2737-2740.
[
Abstract
]
[
Full
Text
]
Read more
GPS-PUP
Recent experiments revealed the prokaryotic ubiquitin-like protein (PUP) to be a signal for the selective
degradation of proteins in Mycobacterium tuberculosis (Mtb).
By covalently conjugating the PUP, pupylation functions as a critical post-translational modification (PTM)
conserved in actinomycetes.
Here, we designed a novel computational tool of GPS-PUP for the prediction of pupylation sites, which was
shown to have a promising performance.
From small-scale and large-scale studies we collected 238 potentially pupylated substrates for which the
exact pupylation sites were still not determined.
As an example application, we predicted ∼85% of these proteins with at least one potential pupylation site.
Furthermore, through functional analysis,
we observed that pupylation can target various substrates so as to regulate a broad array of biological
processes, such as the response to stress, sulfate and proton transport, and metabolism.
The GPS-PUP 1.0 is freely available at:
http://pup.biocuckoo.org
Computational Analysis of Phosphoproteomics: Progresses and Perspectives
Current Protein & Peptide Science. 2011;7(12):591-601.
[
Abstract
]
[
Full Text
]
Read more
Phosphoproteomics
Phosphorylation is one of the most essential post-translational modifications (PTMs) of proteins, regulates
a variety of cellular signaling pathways, and at least partially determines the biological diversity. Recent
progresses in phosphoproteomics have identified more than 100,000 phosphorylation sites, while this number
will easily exceed one million in the next decade. In this regard, how to extract useful information from
flood of phosphoproteomics data has emerged as a great challenge. In this review, we summarized the leading
edges on computational analysis of phosphoproteomics, including discovery of phosphorylation motifs from
phosphoproteomics data, systematic modeling of phosphorylation network, analysis of genetic variation that
influences phosphorylation, and phosphorylation evolution. Based on existed knowledge, we also raised
several perspectives for further studies. We believe that integration of experimental and computational
analyses will propel the phosphoproteomics research into a new phase.
Systematic Analysis of Protein Phosphorylation Networks From Phosphoproteomic Data
Molecular & Cellular Proteomics. 2012;11(10):1070-1083.
[
Abstract
]
[
Full Text
]
Read more
iGPS
In eukaryotes, hundreds of protein kinases (PKs) specifically and precisely modify thousands of substrates
at specific amino
acid residues to faithfully orchestrate numerous biological processes, and reversibly determine the cellular
dynamics and
plasticity. Although over 100,000 phosphorylation sites (p-sites) have been experimentally identified from
phosphoproteomic
studies, the regulatory PKs for most of these sites still remain to be characterized. Here, we present a
novel software package
of iGPS for the prediction of in vivo site-specific kinase-substrate relations mainly from the
phosphoproteomic data.By critical evaluations and comparisons,
the performance of iGPS is satisfying and better than other existed tools. Based on the prediction results,
we modeled protein
phosphorylation networks and observed that the eukaryotic phospho-regulation is poorly conserved at the site
and substrate
levels.This work contributes to the understanding of phosphorylation mechanisms at the systemic level, and
provides a powerful methodology for the general analysis of in vivo post-translational
modifications regulating sub-proteomes.

Systematic analysis of the Plk-mediated phosphoregulation in eukaryotes
Briefings in Bioinformatics. 2013;14(3):344-360.
[
Abstract
]
[
Full Text
]
Read more
Plk-mediated phosphoregulation
Substantial evidence has confirmed that Polo-like kinases (Plks) play a crucial role in a variety of
cellular processes via phosphorylation-mediated signaling transduction.
Identification of Plk phospho-binding proteins and phosphorylation substrates is fundamental for elucidating
the molecular mechanisms of Plks.
Here, we present an integrative approach for the analysis of Plk-specific phospho-binding and
phosphorylation sites (p-sites) in proteins.
From the currently available phosphoproteomic data, we predicted tens of thousands of potential Plk
phospho-binding and phosphorylation sites in eukaryotes, respectively.
Furthermore, statistical analysis suggested that Plk phospho-binding proteins are more closely implicated in
mitosis than their phosphorylation substrates.
Additional computational analysis together with in vitro and in vivo experimental assays demonstrated that
human Mis18B is a novel interacting partner of Plk1, while pT14 and pS48 of Mis18B were identified as
phospho-binding sites.
Taken together, this systematic analysis provides a global landscape of the complexity and diversity of
potential Plk-mediated phosphoregulation, and the prediction results can be helpful for further experimental
investigation.
GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs
Nucleic Acids Research. 2014;42: W325-30.
[
Abstract
]
[
Full
Text
]
ESI HCP
Read more
GPS-SUMO 2.0
Small ubiquitin-like modifiers (SUMOs) regulate a variety of cellular processes through two distinct
mechanisms, including covalent sumoylation and noncovalent SUMO interaction. The complexity of SUMO
regulations has greatly hampered the large-scale identification of SUMO substrates or interaction partners
on a proteome-wide level. In this work, we developed a new tool called GPS-SUMO for the prediction of both
sumoylation sites and SUMO-interaction motifs (SIMs) in proteins. To obtain an accurate performance, a new
generation group-based prediction system (GPS) algorithm integrated with Particle Swarm Optimization
approach was applied. By critical evaluation and comparison, GPS-SUMO was demonstrated to be substantially
superior against other existing tools and methods. With the help of GPS-SUMO, it is now possible to further
investigate the relationship between sumoylation and SUMO interaction processes. A web service of GPS-SUMO
was implemented in PHP + JavaScript and freely available at
http://sumosp.biocuckoo.org.

An integrated overview of spatiotemporal organization and regulation in mitosis in terms of the proteins in
the functional supercomplexes
Frontiers in Microbiology. 2014;5:573.
[
Abstract
]
[
Full Text
]
Read more
Overview
Eukaryotic cells may divide via the critical cellular process of cell division/mitosis, resulting in two
daughter cells with the same genetic information. A large number of dedicated proteins are involved in this
process and spatiotemporally assembled into three distinct super-complex structures/organelles, including
the centrosome/spindle pole body, kinetochore/centromere and cleavage furrow/midbody/bud neck, so as to
precisely modulate the cell division/mitosis events of chromosome alignment, chromosome segregation and
cytokinesis in an orderly fashion. In recent years, many efforts have been made to identify the protein
components and architecture of these subcellular organelles, aiming to uncover the organelle assembly
pathways, determine the molecular mechanisms underlying the organelle functions, and thereby provide new
therapeutic strategies for a variety of diseases. However, the organelles are highly dynamic structures,
making it difficult to identify the entire components. Here, we review the current knowledge of the
identified protein components governing the organization and functioning of organelles, especially in human
and yeast cells, and discuss the multi-localized protein components mediating the communication between
organelles during cell division.

IBS: an illustrator for the presentation and visualization of biological sequences
Bioinformatics. 2015;31(20):3359-61.
[
Abstract
]
[
Full Text
]
ESI Hot Paper
Read more
IBS 1.0
Biological sequence diagrams are fundamental for visualizing various functional elements in protein or
nucleotide sequences that enable a summarization and presentation of existing information as well as means
of intuitive new discoveries. Here, we present a software package called illustrator of biological sequences
(IBS) that can be used for representing the organization of either protein or nucleotide sequences in a
convenient, efficient and precise manner. Multiple options are provided in IBS, and biological sequences can
be manipulated, recolored or rescaled in a user-defined mode. Also, the final representational artwork can
be directly exported into a publication-quality figure.
The standalone package of IBS was implemented in JAVA, while the online service was implemented in HTML5 and
JavaScript. Both the standalone package and online service are freely available at
http://ibs.biocuckoo.org.
RPFdb: a database for genome wide information of translated mRNA generated from ribosome profiling.
Nucleic Acids Research. 2016;44:D254-D258.
[
Abstract
]
[
Full Text
]
Read more
RPFdb
Translational control is crucial in the regulation of gene expression and deregulation of translation is
associated with a wide range of cancers and human diseases. Ribosome profiling is a technique that provides
genome wide information of mRNA in translation based on deep sequencing of ribosome protected mRNA fragments
(RPF). RPFdb is a comprehensive resource for hosting, analyzing and visualizing RPF data, available at
http://www.rpfdb.org. The current version
of database contains 777 samples from 82 studies in 8 species, processed and reanalyzed by a unified
pipeline. Overall our database provides a simple way to search, analyze, compare, visualize and download RPF
data sets.

GPS-Lipid: a robust tool for the prediction of multiple lipid modification sites
Scientific Reports. 2016;6:28249.
[
Abstract
]
[
Full Text
]
Read more
GPS-Lipid 1.0
As one of the most common post-translational modifications in eukaryotic cells, lipid modification is an
important mechanism for the regulation of variety aspects of protein function. In this work, we developed a
tool called GPS-Lipid for the prediction of four classes of lipid modifications by integrating the Particle
Swarm Optimization with an aging leader and challengers (ALC-PSO) algorithm. GPS-Lipid was proven to be
evidently superior to other similar tools. To facilitate the research of lipid modification, we hosted a
publicly available web server at
http://lipid.biocuckoo.org
with not only the implementation of GPSLipid, but also an integrative database and visualization tool. We
performed a systematic analysis of the co-regulatory mechanism between different lipid modifications with
GPS-Lipid. The results demonstrated that the proximal dual-lipid modifications among palmitoylation,
myristoylation and prenylation are key mechanism for regulating various protein functions. In conclusion,
GPS-lipid is expected to serve as useful resource for the research on lipid modifications, especially on
their coregulation.

VirusMap: A visualization database for the influenza A virus
Journal of Genetics and Genomics. 2017;44(4):281-284.
[
Abstract
]
[
Full Text
]
Read more
VirusMap
In this study, we reported a visualization platform called VirusMap, which is available at the website (
http://virusmap.renlab.org), for
investigating the epidemiological and geographical distribution of influenza A viruses. We downloaded
615,866 protein and 482,663 nucleotide sequences of influenza A viruses in FASTA format from IVR(Bao et al.,
2008) andIRD(Squires et al., 2012). As the policy for the data submission in those databases, the
information of subtype, host, sampling location, sampling time and serotype should be included for each
virus strain. Thus, the title line of each FASTA sequence contains all of the necessary information. We
extracted these information through a semi-automated series of steps. To ensure the data quality, only
entries with the full information of host, serotype and sampling information were preserved. In total, there
were 583,052 protein and 448,495nucleotide records retained in a MySQL database. As the data were obtained
from the two most popular influenza virus resources, VirusMap contains a comprehensive and frequently
updated dataset on the influenza A virus.

Firmiana: towards a one-stop proteomic cloud platform for data processing and analysis
Nature Biotechnology. 2017;35:409–412.
[
Abstract
]
[
Full Text
]
Read more
Firmiana
Improvements in next-generation proteomics, including instrumentation, sample preparation, and computational
analysis, have generated large amounts of data that cover protein profiling, post-translational
modifications, and protein–protein interactions. The first draft of the human proteome, for example, made
use of 2,000 (ref. 6) and 16,000 (ref. 5) raw files. Proteomics now calls for a uniform online pipeline that
can host millions of data sets with the same quality standards, analyze hundreds to thousands of
experiments, and integrate multi-dimensional omics data for knowledge mining and hypothesis generation to
disseminate proteomics to the scientific community. Here, we describe Firmiana (V1.0) (
http://www.firmiana.org/), a one-stop proteomic data processing and
integrated omics analysis cloud platform that allows scientists to deposit mass spectrometry (MS) raw files,
perform proteome identification and quantification online, carry out bioinformatics analyses, extract
knowledge, and visualize results using a biologist-friendly web interface without the need for programming
expertise.
A de novo substructure generation algorithm for identifying the privileged chemical fragments of liver X
receptorβ agonists
Scientific Reports. 2017;7:11121.
[
Abstract
]
[
Full Text
]
Read more
Overview
Liver X receptorβ (LXRβ) is a promising therapeutic target for lipid disorders, atherosclerosis, chronic
inflammation, autoimmunity, cancer and neurodegenerative diseases. Druggable LXRβ agonists have been
explored over the past decades. However, the pocket of LXRβ ligand-binding domain (LBD) is too large to
predict LXRβ agonists with novel scaffolds based on either receptor or agonist structures. In this paper, we
report a de novo algorithm which drives privileged LXRβ agonist fragments by starting with individual
chemical bonds (de novo) from every molecule in a LXRβ agonist library, growing the bonds into substructures
based on the agonist structures with isomorphic and homomorphic restrictions, and electing the privileged
fragments from the substructures with a popularity threshold and background chemical and biological
knowledge. Using these privileged fragments as queries, we were able to figure out the rules to reconstruct
LXRβ agonist molecules from the fragments. The privileged fragments were validated by building regularized
logistic regression (RLR) and supporting vector machine (SVM) models as descriptors to predict a LXRβ
agonist activities.

m6AVar: a database of functional variants involved in m6A modification.
Nucleic Acids Research. 2018; 46(D1): D139-145.
[
Abstract
]
[
Full Text
]
Read more
m6AVar
Here, we report m6AVar (
http://m6avar.renlab.org), a comprehensive
database of m6A-associated variants that potentially influence m6A modification, which will help to
interpret variants by m6A function. The m6A-associated variants were derived from three different m6A
sources including miCLIP/PA-m6A-seq experiments (high confidence), MeRIP-Seq experiments (medium confidence)
and transcriptome-wide predictions (low confidence). Currently, m6AVar contains 16,132 high, 71,321 medium
and 326,915 low confidence level m6A-associated variants. We also integrated the RBP-binding regions,
miRNA-targets and splicing sites associated with variants to help users investigate the effect of
m6A-associated variants on post-transcriptional regulation. Because it integrates the data from genome-wide
association studies (GWAS) and ClinVar, m6AVar is also a useful resource for investigating the relationship
between the m6A-assocaited variants and disease. Overall, m6AVar will serve as a useful resource for
annotating variants and identifying disease-causing variants.
Expression and regulation of long noncoding RNAs during the osteogenic differentiation of periodontal ligament
stem cells in the inflammatory microenvironment
Scientific Reports. 2017;7:13991.
[
Abstract
]
[
Full Text
]
Read more
Overview
Although long noncoding RNAs (lncRNAs) have been emerging as critical regulators in various tissues and
biological processes, little is known about their expression and regulation during the osteogenic
differentiation of periodontal ligament stem cells (PDLSCs) in inflammatory microenvironment. In this study,
we have identified 63 lncRNAs that are not annotated in previous database. These novel lncRNAs were not
randomly located in the genome but preferentially located near protein-coding genes related to particular
functions and diseases, such as stem cell maintenance and differentiation, development disorders and
inflammatory diseases. Moreover, we have identified 650 differentially expressed lncRNAs among different
subsets of PDLSCs. Pathway enrichment analysis for neighboring protein-coding genes of these differentially
expressed lncRNAs revealed stem cell differentiation related functions. Many of these differentially
expressed lncRNAs function as competing endogenous RNAs that regulate protein-coding transcripts through
competing shared miRNAs.

Read more
m6ASNP
Background: Large-scale genome sequencing projects have identified many genetic variants for diverse
diseases. A major goal of these projects is to characterize these genetic variants to provide insight into
their function and roles in diseases. N6-methyladenosine (m6A) is one of the most abundant RNA modifications
in eukaryotes. Recent studies have revealed that aberrant m6A modifications are involved in many
diseases.
Findings: In this study, we present a user-friendly web server called “m6ASNP” that is dedicated to
the identification of genetic variants targeting m6A modification sites. A random forest model was
implemented in m6ASNP to predict whether the methylation status of a m6A site is altered by the variants
surrounding the site. In m6ASNP, genetic variants in a standard VCF format are accepted as the input data,
and the output includes an interactive table containing the genetic variants annotated by m6A function. In
addition, statistical diagrams and a genome browser are provided to visualize the characteristics and
annotate the genetic variants.
Conclusions: We believe that m6ASNP is a highly convenient tool that can be used to boost further
functional studies investigating genetic variants. The web server “m6ASNP” is implemented in JAVA and PHP
and is freely available at http://m6asnp.renlab.org.

Pan-Cancer Analysis Reveals the Functional Importance of Protein Lysine Modification in Cancer
Development
Front. Genet. 9:254. doi: 10.3389/fgene.2018.00254
[
Abstract
]
[
Full Text
]
Read more
Overview
Large-scale tumor genome sequencing projects have revealed a complex landscape of genomic
mutations in multiple cancer types. A major goal of these projects is to characterize somatic
mutations and discover cancer drivers, thereby providing important clues to uncover diagnostic
or therapeutic targets for clinical treatment. However, distinguishing only a few somatic
mutations from the majority of passenger mutations is still a major challenge facing the
biological community. Fortunately, combining other functional features with mutations to
predict cancer driver genes is an effective approach to solve the above problem. Protein
lysine modifications are an important functional feature that regulates the development of
cancer. Therefore, in this work, we have systematically analyzed somatic mutations on seven
protein lysine modifications and identified several important drivers that are responsible for
tumorigenesis. From published literature, we first collected more than 100,000 lysine
modification sites for analysis. Another 1 million non-synonymous single nucleotide variants
(SNVs) were then downloaded from TCGA and mapped to our collected lysine modification sites.
To identify driver proteins that significantly altered lysine modifications, we further
developed a hierarchical Bayesian model and applied the Markov Chain Monte Carlo (MCMC) method
for testing. Strikingly, the coding sequences of 473 proteins were found to carry a higher
mutation rate in lysine modification sites compared to other background regions.
Hypergeometric tests also revealed that these gene products were enriched in known cancer
drivers. Functional analysis suggested that mutations within the lysine modification regions
possessed higher evolutionary conservation and deleteriousness. Furthermore, pathway
enrichment showed that mutations on lysine modification sites mainly affected cancer related
processes, such as cell cycle and RNA transport. Moreover, clinical studies also suggested
that the driver proteins were significantly associated with patient survival, implying an
opportunity to use lysine modifications as molecular markers in cancer diagnosis or treatment.
By searching within protein-protein interaction networks using a random walk with restart
(RWR) algorithm, we further identified a series of potential treatment agents and therapeutic
targets for cancer related to lysine modifications. Collectively, this study reveals the
functional importance of lysine modifications in cancer development and may benefit the
discovery of novel mechanisms for cancer treatment.
m6A RNA modification controls autophagy through upregulating ULK1 protein abundance
Cell Research. 2018;
[
Abstract
]
[
Full Text
]
Read more
Overview
N6-methyladenosine (m6A) is the prominent dynamic mRNA modification, governed
by methyltransferase complex (“writers”), demethylases (“erasers”) and RNA-binding
proteins (‘readers’).1 m6A modification directs mRNAs to distinct fates by grouping them for
differential processing, translation and decay in the processes such as cell differentiation,
embryonic development and stress responses. Owing to a deeper understanding of this
modification and the technological advance, functional characterizations of m6A in gene
regulation have become a hot topic that warrants further dissection.

DeepNitro: Prediction of Protein Nitration and Nitrosylation Sites by Deep Learning
Genomics Proteomics Bioinformatics. 2018; 16(4): 294-306.
[
Abstract
]
[
Full Text
]
Read more
DeepNitro
Protein nitration and nitrosylation are essential post-translational modifications (PTMs) involved in many
fundamental cellular processes. Recent studies have revealed that excessive levels of nitration and nitrosylation
in some critical proteins are linked to numerous chronic diseases. Therefore, the identification of substrates
that undergo such modifications in a site-specific manner is an important research topic in the community and will
provide candidates for targeted therapy. In this study, we aimed to develop a computational tool for predicting
nitration and nitrosylation sites in proteins. We first constructed four types of encoding features, including
positional amino acid distributions, sequence contextual dependencies, physicochemical properties, and
position-specific scoring features, to represent the modified residues. Based on these encoding features, we
established a predictor called DeepNitro using deep learning methods for predicting protein nitration and
nitrosylation. Using n-fold cross-validation, our evaluation shows great AUC values for DeepNitro, 0.65 for
tyrosine nitration, 0.80 for tryptophan nitration, and 0.70 for cysteine nitrosylation, respectively,
demonstrating the robustness and reliability of our tool. Also, when tested in the independent dataset, DeepNitro
is substantially superior to other similar tools with a 7%−42% improvement in the prediction performance. Taken
together, the application of deep learning method and novel encoding schemes, especially the position-specific
scoring feature, greatly improves the accuracy of nitration and nitrosylation site prediction and may facilitate
the prediction of other PTM sites. DeepNitro is implemented in JAVA and PHP and is freely available for academic
research at
http://deepnitro.renlab.org.

lnCAR: a comprehensive resource for lncRNAs from Cancer Arrays.
Cancer Res February 20 2019 DOI: 10.1158/0008-5472.CAN-18-2169
[
Abstract
]
[
Full
Text
]
Read more
lnCAR
Long non-coding RNAs (lncRNA) have emerged as promising biomarkers in cancer diagnosis, treatment, and prognosis.
Recent studies suggest that a large number of coding gene expression microarray probes could be re-annotated as
lncRNAs. Microarray, once the most cutting-edge high throughput gene expression technology, has been used for
thousands of cancer studies and has brought invaluable resources for studying the functions of lncRNA in cancer
development. However, a comprehensive lncRNA resource based on microarray data is still lacking. Here we present
lnCAR, a comprehensive open resource for providing expression profiles and prognostic landscape of lncRNAs derived
from re-annotation of public microarray data. Currently, lnCAR contains 52,300 samples for differential expression
analysis and 12,883 samples for survival analysis from 10 cancer types. lnCAR allows users to interactively
explore any annotated or novel lncRNAs. We believe lnCAR will serve as a valuable resource for the community
focused on lncRNA research in cancer.

DeepPhagy: a deep learning framework for quantitatively measuring autophagy activity in Saccharomyces
cerevisiae.
Autophagy. Jun 12 2019 DOI: 10.1080/15548627.2019.1632622
[
Abstract
]
[
Full
Text
]
Read more
DeepPhagy
Seeing is believing. The direct observation of GFP-Atg8 vacuolar delivery under confocal microscopy is one of the
most useful end-point measurements for monitoring yeast macroautophagy/autophagy. However, manually labelling
individual cells from large-scale sets of images is time-consuming and labor-intensive, which has greatly hampered
its extensive use in functional screens. Herein, we conducted a time-course analysis of nitrogen
starvation-induced autophagy in wild-type and knockout mutants of 35 AuTophaGy-related (ATG) genes in
Saccharomyces cerevisiae and obtained 1,944 confocal images containing > 200,000 cells. We manually labelled 8,078
autophagic and 18,493 non-autophagic cells as a benchmark dataset and developed a new deep learning tool for
autophagy (DeepPhagy), which exhibited superior accuracy in recognizing autophagic cells compared to other
existing methods, with an area under the curve (AUC) value of 0.9710 from 10-fold cross-validations. We further
used DeepPhagy to automatically analyze all the images and quantitatively classified the autophagic phenotypes of
the 35 atg knockout mutants into 3 classes. The high consistency in our computational and biochemical results
indicated the reliability of DeepPhagy for measuring autophagic activity. Moreover, we used DeepPhagy to analyze 3
additional types of autophagic phenotypes, including the targeting of Atg1-GFP to the vacuole, the vacuolar
delivery of GFP-Atg19, and the disintegration of autophagic bodies indicated by GFP-Atg8, all with satisfying
accuracies. Taken together, our study not only enables the GFP-Atg8 fluorescence assay to become a quantitative
measurement for analyzing autophagic phenotypes in S. cerevisiae but also demonstrates that deep learning-based
methods could potentially be applied to different types of autophagy.
BBCancer: an expression atlas of blood-based biomarkers in the early diagnosis of cancers
Nucleic Acids Research. October 29 2019 DOI: 10.1093/nar/gkz942
[
Abstract
]
[
Full Text
]
Read more
BBCancer
The early detection of cancer holds the key to combat and control the increasing global burden of cancer morbidity and mortality. Blood-based screenings using circulating DNAs (ctDNAs), circulating RNA (ctRNAs), circulating tumor cells (CTCs) and extracellular vesicles (EVs) have shown promising prospects in the early detection of cancer. Recent high-throughput gene expression profiling of blood samples from cancer patients has provided a valuable resource for developing new biomarkers for the early detection of cancer. However, a well-organized online repository for these blood-based high-throughput gene expression data is still not available. Here, we present BBCancer (http://bbcancer.renlab.org/), a web-accessible and comprehensive open resource for providing the expression landscape of six types of RNAs, including messenger RNAs (mRNAs), long noncoding RNAs (lncRNAs), microRNAs (miRNAs), circular RNAs (circRNAs), tRNA-derived fragments (tRFRNAs) and Piwi-interacting RNAs (piRNAs) in blood samples, including plasma, CTCs and EVs, from cancer patients with various cancer types. Currently, BBCancer contains expression data of the six RNA types from 5040 normal and tumor blood samples across 15 cancer types. We believe this database will serve as a powerful platform for developing blood biomarkers.
RMVar: an updated database of functional variants involved in RNA modifications
Nucleic Acids Research. 06 October 2020 DOI: 10.1093/nar/gkaa811
[
Abstract
]
[
Full Text
]
Read more
RMVar
Distinguishing the few disease-related variants from a massive number of passenger variants is a major challenge.
Variants affecting RNA modifications that play critical roles in many aspects of RNA metabolism have recently been linked to many human diseases,
such as cancers. Evaluating the effect of genetic variants on RNA modifications will provide a new perspective for understanding the pathogenic mechanism of human diseases.
Previously, we developed a database called ‘m6AVar’ to host variants associated with m6A, one of the most prevalent RNA modifications in eukaryotes.
To host all RNA modification (RM)-associated variants, here we present an updated version of m6AVar renamed RMVar (http://rmvar.renlab.org). In this update,
RMVar contains 1 678 126 RM-associated variants for 9 kinds of RNA modifications, namely m6A, m6Am, m1A, pseudouridine, m5C, m5U, 2′-O-Me, A-to-I and m7G,
at three confidence levels. Moreover, RBP binding regions, miRNA targets, splicing events and circRNAs were integrated to assist investigations of the effects of
RM-associated variants on posttranscriptional regulation. In addition, disease-related information was integrated from ClinVar and other genome-wide association studies (GWAS) to
investigate the relationship between RM-associated variants and diseases. We expect that RMVar may boost further functional studies on genetic variants affecting RNA modifications.

PTMsnp: A Web Server for the Identification of Driver Mutations That Affect Protein Post-translational Modification
Frontiers in Cell and Developmental Biology. 10 November 2020 DOI: 10.3389/fcell.2020.593661
[
Abstract
]
[
Full Text
]
Read more
PTMsnp
High-throughput sequencing technologies have identified millions of genetic mutations in multiple human diseases. However, the interpretation of the pathogenesis of these mutations and the discovery of driver genes that dominate disease progression is still a major challenge. Combining functional features such as protein post-translational modification (PTM) with genetic mutations is an effective way to predict such alterations. Here, we present PTMsnp, a web server that implements a Bayesian hierarchical model to identify driver genetic mutations targeting PTM sites. PTMsnp accepts genetic mutations in a standard variant call format or tabular format as input and outputs several interactive charts of PTM-related mutations that potentially affect PTMs. Additional functional annotations are performed to evaluate the impact of PTM-related mutations on protein structure and function, as well as to classify variants relevant to Mendelian disease. A total of 4,11,574 modification sites from 33 different types of PTMs and 1,776,848 somatic mutations from TCGA across 33 different cancer types are integrated into the web server, enabling identification of candidate cancer driver genes based on PTM. Applications of PTMsnp to the cancer cohorts and a GWAS dataset of type 2 diabetes identified a set of potential drivers together with several known disease-related genes, indicating its reliability in distinguishing disease-related mutations and providing potential molecular targets for new therapeutic strategies. PTMsnp is freely available at: http://ptmsnp.renlab.org.

autoRPA: A web server for constructing cancer staging models by recursive partitioning analysis
Computational and Structural Biotechnology Journal. 10 November 2020 DOI: 10.1016/j.csbj.2020.10.038
[
Abstract
]
[
Full Text
]
Read more
autoRPA
Cancer staging provides a common language that is used to describe the severity of an individual's cancer, which plays a critical role in optimizing cancer treatment. Recursive partitioning analysis (RPA) is the most widely accepted method for cancer staging. Despite its widespread use, to date, only limited tools have been developed to implement the RPA algorithm for cancer staging. Moreover, most of the available tools can be accessed only from command lines and also lack visualization, making them difficult for clinical investigators without programing skills to use. Therefore, we developed a web server called autoRPA that is dedicated to supporting the construction of prognostic staging models and performance comparisons among different staging models. Based on the RPA algorithm and log-rank test statistics, autoRPA can establish a decision-making tree from survival data and provide clinicians an intuitive method to further prune the decision tree. Moreover, autoRPA can evaluate the contribution of each submitted covariate that is involved in the grouping process and help identify factors that significantly contribute to cancer staging. Four indicators, including hazard consistency, hazard discrimination, percentage of variation explained, and sample size balance, are introduced to validate the performance of the designed staging models. In addition, autoRPA can also be used to compare the performance of different prognostic staging models using a standard bootstrap evaluation method. The web server of autoRPA is freely available at http://rpa.renlab.org.
DeepOMe: A Web Server for the Prediction of 2′-O-Me Sites Based on the Hybrid CNN and BLSTM Architecture
Frontiers in Cell and Developmental Biology. 14 May 2021 DOI: 10.3389/fcell.2021.686894
[
Abstract
]
[
Full Text
]
Read more
DeepOMe
2′-O-methylations (2′-O-Me or Nm) are one of the most important layers of regulatory control over gene expression. With increasing attentions focused on the characteristics, mechanisms and influences of 2′-O-Me, a revolutionary technique termed Nm-seq were established, allowing the identification of precise 2′-O-Me sites in RNA sequences with high sensitivity. However, as the costs and complexities involved with this new method, the large-scale detection and in-depth study of 2′-O-Me is still largely limited. Therefore, the development of a novel computational method to identify 2′-O-Me sites with adequate reliability is urgently needed at the current stage. To address the above issue, we proposed a hybrid deep-learning algorithm named DeepOMe that combined Convolutional Neural Networks (CNN) and Bidirectional Long Short-term Memory (BLSTM) to accurately predict 2′-O-Me sites in human transcriptome. Validating under 4-, 6-, 8-, and 10-fold cross-validation, we confirmed that our proposed model achieved a high performance (AUC close to 0.998 and AUPR close to 0.880). When testing in the independent data set, DeepOMe was substantially superior to NmSEER V2.0. To facilitate the usage of DeepOMe, a user-friendly web-server was constructed, which can be freely accessed at http://deepome.renlab.org.
MesKit: a tool kit for dissecting cancer evolution of multi-region tumor biopsies through somatic alterations
GigaScience. 21 May 2021 DOI: 10.1093/gigascience/giab036
[
Abstract
]
[
Full Text
]
Read more
MesKit
Multi-region sequencing (MRS) has been widely used to analyze intra-tumor heterogeneity (ITH) and cancer evolution. However, comprehensive analysis of mutational data from MRS is still challenging, necessitating complicated integration of a plethora of computational and statistical approaches.
Here, we present MesKit, an R/Bioconductor package that can assist in characterizing genetic ITH and tracing the evolutionary history of tumors based on somatic alterations detected by MRS. MesKit provides a wide range of analysis and visualization modules, including ITH evaluation, metastatic route inference, and mutational signature identification. In addition, MesKit implements an auto-layout algorithm to generate phylogenetic trees based on somatic mutations. The application of MesKit for 2 reported MRS datasets of hepatocellular carcinoma and colorectal cancer identified known heterogeneous features and evolutionary patterns, together with potential driver events during cancer evolution.
In summary, MesKit is useful for interpreting ITH and tracing evolutionary trajectory based on MRS data. MesKit is implemented in R and available at https://bioconductor.org/packages/MesKit under the GPL v3 license.
SPENCER: a comprehensive database for small peptides encoded by noncoding RNAs in cancer patients
Nucleic Acids Research. 27 September 2021 DOI: 10.1093/nar/gkab822
[
Abstract
]
[
Full Text
]
Read more
SPENCER
As an increasing number of noncoding RNAs (ncRNAs) have been suggested to encode short bioactive peptides in cancer, the exploration of ncRNA-encoded small peptides (ncPEPs) is emerging as a fascinating field in cancer research. To assist in studies on the regulatory mechanisms of ncPEPs, we describe here a database called SPENCER (http://spencer.renlab.org). Currently, SPENCER has collected a total of 2806 mass spectrometry (MS) data points from 55 studies, covering 1007 tumor samples and 719 normal samples. Using an MS-based proteomics analysis pipeline, SPENCER identified 29 526 ncPEPs across 15 different cancer types. Specifically, 22 060 of these ncPEPs were experimentally validated in other studies. By comparing tumor and normal samples, the identified ncPEPs were divided into four expression groups: tumor-specific, upregulated in cancer, downregulated in cancer, and others. Additionally, since ncPEPs are potential targets for neoantigen-based cancer immunotherapy, SPENCER also predicted the immunogenicity of all the identified ncPEPs by assessing their MHC-I binding affinity, stability, and TCR recognition probability. As a result, 4497 ncPEPs curated in SPENCER were predicted to be immunogenic. Overall, SPENCER will be a useful resource for investigating cancer-associated ncPEPs and may boost further research in cancer.
RPS: a comprehensive database of RNAs involved in liquid–liquid phase separation
Nucleic Acids Research. 28 October 2021 DOI: 10.1093/nar/gkab986
[
Abstract
]
[
Full Text
]
Read more
RPS
Liquid–liquid phase separation (LLPS) is critical for assembling membraneless organelles (MLOs) such as nucleoli, P-bodies, and stress granules, which are involved in various physiological processes and pathological conditions. While the critical role of RNA in the formation and the maintenance of MLOs is increasingly appreciated, there is still a lack of specific resources for LLPS-related RNAs. Here, we presented RPS (http://rps.renlab.org), a comprehensive database of LLPS-related RNAs in 20 distinct biomolecular condensates from eukaryotes and viruses. Currently, RPS contains 21,613 LLPS-related RNAs with three different evidence types, including ‘Reviewed’, ‘High-throughput’ and ‘Predicted’. RPS provides extensive annotations of LLPS-associated RNA properties, including sequence features, RNA structures, RNA–protein/RNA–RNA interactions, and RNA modifications. Moreover, RPS also provides comprehensive disease annotations to help users to explore the relationship between LLPS and disease. The user-friendly web interface of RPS allows users to access the data efficiently. In summary, we believe that RPS will serve as a valuable platform to study the role of RNA in LLPS and further improve our understanding of the biological functions of LLPS.
TIRSF: a web server for screening gene signatures to predict Tumor immunotherapy response
Nucleic Acids Research. 12 May 2022 DOI: 10.1093/nar/gkac374
[
Abstract
]
[
Full Text
]
Read more
TIRSF
Immune checkpoint blockade (ICB) therapy has been successfully applied to clinically therapeutics in multiple cancers, but its efficacy varies greatly among different patients and cancer types. Therefore, the construction of gene signatures to identify patients who could benefit from ICB therapy is particularly important for precision cancer treatment. However, due to the lack of a user-friendly platform, the construction of such gene signatures is a great challenge for clinical investigators who have limited programming skills. In light of this challenge, we developed a web server called Tumor Immunotherapy Response Signature Finder(TIRSF) for the construction of gene signatures to predict ICB therapy response in cancer patients. TIRSF consists of three functional modules. The first module is the Signature Discovery module which provides signature construction and performance evaluation functionalities. The second is a module for response prediction based on the TIRSF signatures, which enables response prediction and prognostic analysis of immunotherapy samples. The last is a module for response prediction based on existing signatures. This module currently integrates 24 published signatures for ICB therapy response prediction. Together, all of above features can be freely accessed at http://tirsf.renlab.org/.
IBS 2.0: an upgraded illustrator for the visualization of biological sequences
Nucleic Acids Research. 17 May 2022 DOI: 10.1093/nar/gkac373
[
Abstract
]
[
Full Text
]
Read more
IBS 2.0
The visualization of biological sequences with various functional elements is fundamental for the publication of scientific achievements in the field of molecular and cellular biology. However, due to the limitations of the currently used applications, there are still considerable challenges in the preparation of biological schematic diagrams. Here, we present a professional tool called IBS 2.0 for illustrating the organization of both protein and nucleotide sequences. With the abundant graphical elements provided in IBS 2.0, biological sequences can be easily represented in a concise and clear way. Moreover, we implemented a database visualization module in IBS 2.0, enabling batch visualization of biological sequences from the UniProt and the NCBI RefSeq databases. Furthermore, to increase the design efficiency, a resource platform that allows uploading, retrieval, and browsing of existing biological sequence diagrams has been integrated into IBS 2.0. In addition, a lightweight JS library was developed in IBS 2.0 to assist the visualization of biological sequences in customized web services. To obtain the latest version of IBS 2.0, please visit https://ibs.renlab.org.

Post-translational Modifications
Our group is engaged in the study of post-translational modifications(PTMs) using computational approaches.
We have
been developing a high-effective algorithm named GPS (Group-based Prediction System) for the
prediction of PTMs sites.
Based on the GPS algorithm,over ten types of PTM predictors have been
released. We also built a series
databases for protein phosphorylation, lipid and lysine modifications.
Recently, we are combining
the computational methods with the technology of BiFC(Bimolecular
Fluorescence
Complementation) to develop a systematic approach for studying
the SUMO regulation in
Homo sapiens.
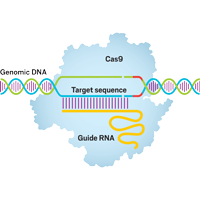
Gene Editting with CRISPR
Our group also focus on developing computational tools for assisting the design of CRISPR system.
Currently, we have
developed a high efficient binary alignment scheme to screen out potential on-target
and off-target sites from
the whole genome. Using machine learning methods, such as Random Forest, we
predicted the cleavage
efficacies of the potential target sites, and recommended an optimal gRNA design
for the users
based on our predictions. A subsequent experimental validation will be also
performed
in the near further.
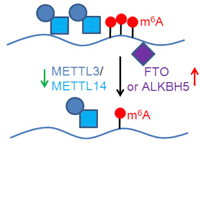
RNA N6-methyladenosine Modification
RNA N6-methyladenosine (m6A) modification has a critical role in the regulation of many fundamental
biological processes.
However, the role of m6A in cancer is poorly understood. We have developed a
computational tool, which is called
“m6A Finder”, for predicting m6A modification sites at
single-nucleotide resolution. We then systematically
investigate the m6A-associated somatic mutations
in cancers using TCGA data. We are also
developing algorithms to analyze m6A-Seq data, such as peak
calling
and differential methylation analysis.